Kasper presented the Stanley Center Brain Interaction Network project to the Open Targets Community. Great job Kasper!
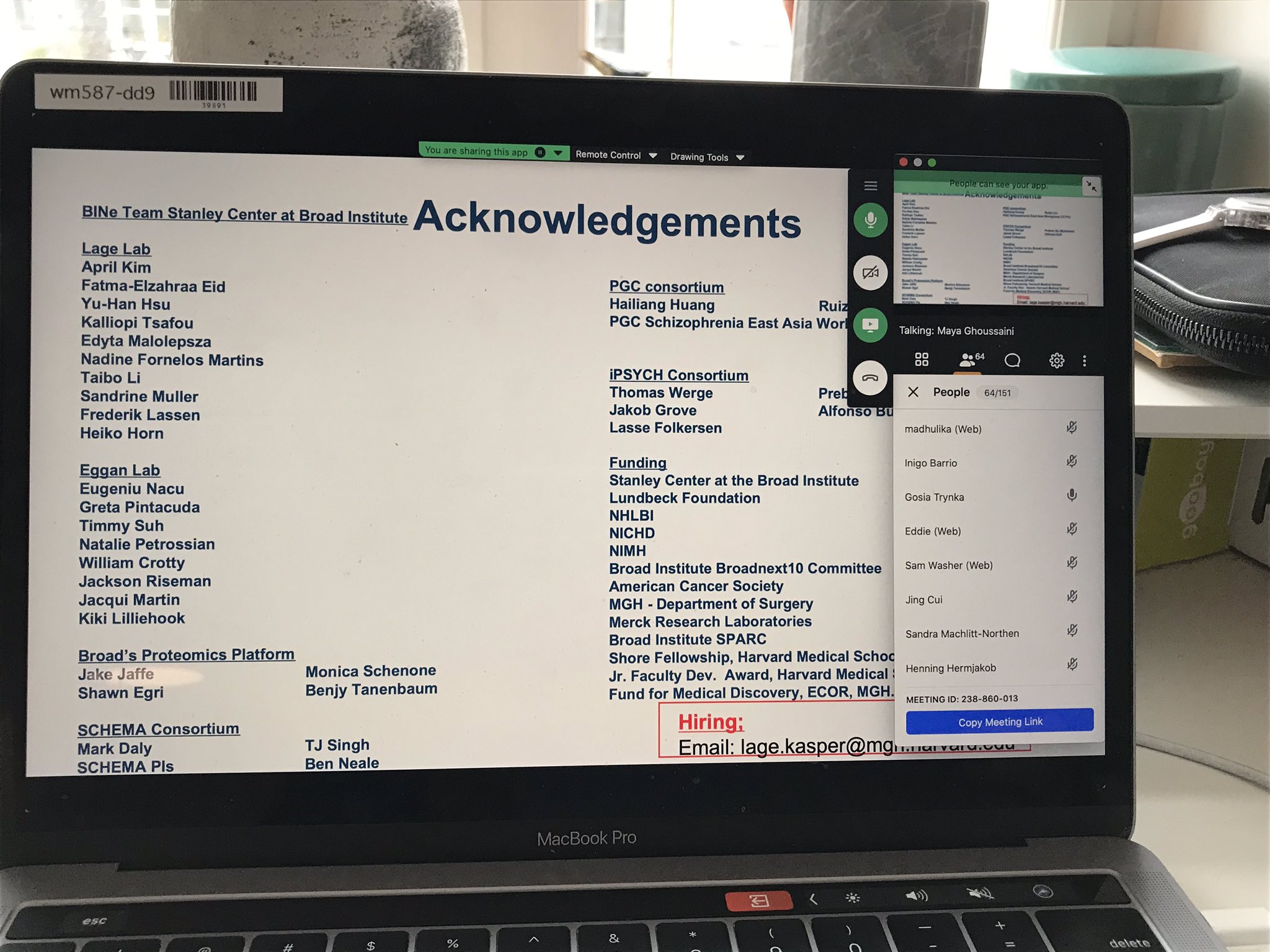
Kasper presented the Stanley Center Brain Interaction Network project to the Open Targets Community. Great job Kasper!
Kasper recently interviewed with Vivien Marx on her Nature Methods Technology Feature titled ‘Bench pressing with genomics benchmarkers,’ which highlights the importance of benchmarking for genomics softwares. The insightful article can be found here: https://www.nature.com/articles/s41592-020-0768-1?proof=trueatural-gas-cut-pollution-saved-lives%2F


Congratulations to Frederik Heymann Lassen on being accepted to the PhD program in Genomics Medicine and Statistics at Oxford University led by Julian Knight and John Todd!
Nadine’s work on identification of new biomarkers of Inflammatory Bowel Disease (IBD), under the direction of Ramnik Xavier in collaboration with the Broad Institute, Harvard School of Public Health, the University of North Carolina and Novartis has been published in Nature Microbiology. Fantastic work Nadine!
Find the digest here: https://naturemicrobiologycommunity.nature.com/users/346662-nadine-fornelos/posts/58611-gut-bugs-and-metabolites.
Find the article here: https://www.nature.com/articles/s41564-019-0655-7

The Brain Interaction Network (BINe) team hosted the February Stanley Center Social and introduced its people to the Stanley Center community.
Kasper hosted the Stanley Center Program Meeting where he gave a talk titled “Functional interpretation of genetic data in psychiatric diseases using human neuronal protein-protein interaction networks.” In addition, Yu-Han Hsu and Greta Pintacuda, postdocs in the Lage Lab and Eggan Lab, presented their work titled “Combining proteomics with human genetics to study neuropsychiatric disease.” Great job everyone!
Poster led by Greta Pintacuda from Kevin Eggan’s lab on our joint Stanley Center at the Broad Institute project to map protein networks of psychiatric risk genes in human neurons won both the popular poster award and judge-based vote at The 2019 Broad Retreat. Only 12 awards were given to 260 featured posters. Congratulations BINe team!
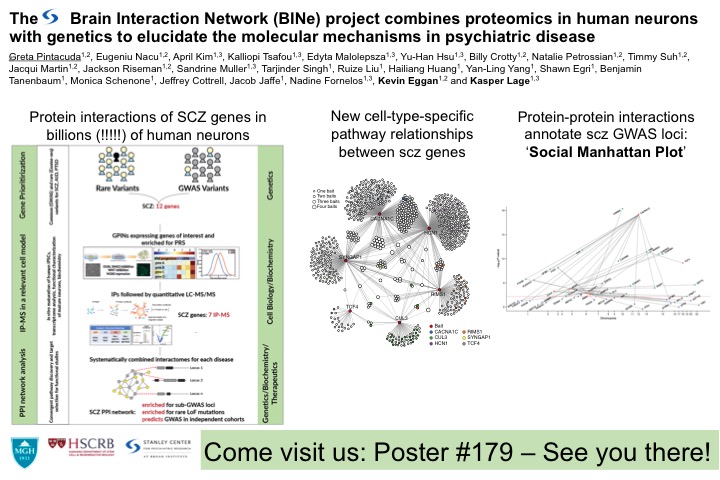

Kasper gave a plenary talk at World Congress of Psychiatric Genetics 2019 where he discussed the functional interpretation of genetic data in psychiatric diseases and the insights from tissue specific protein-protein interaction networks for schizophrenia.

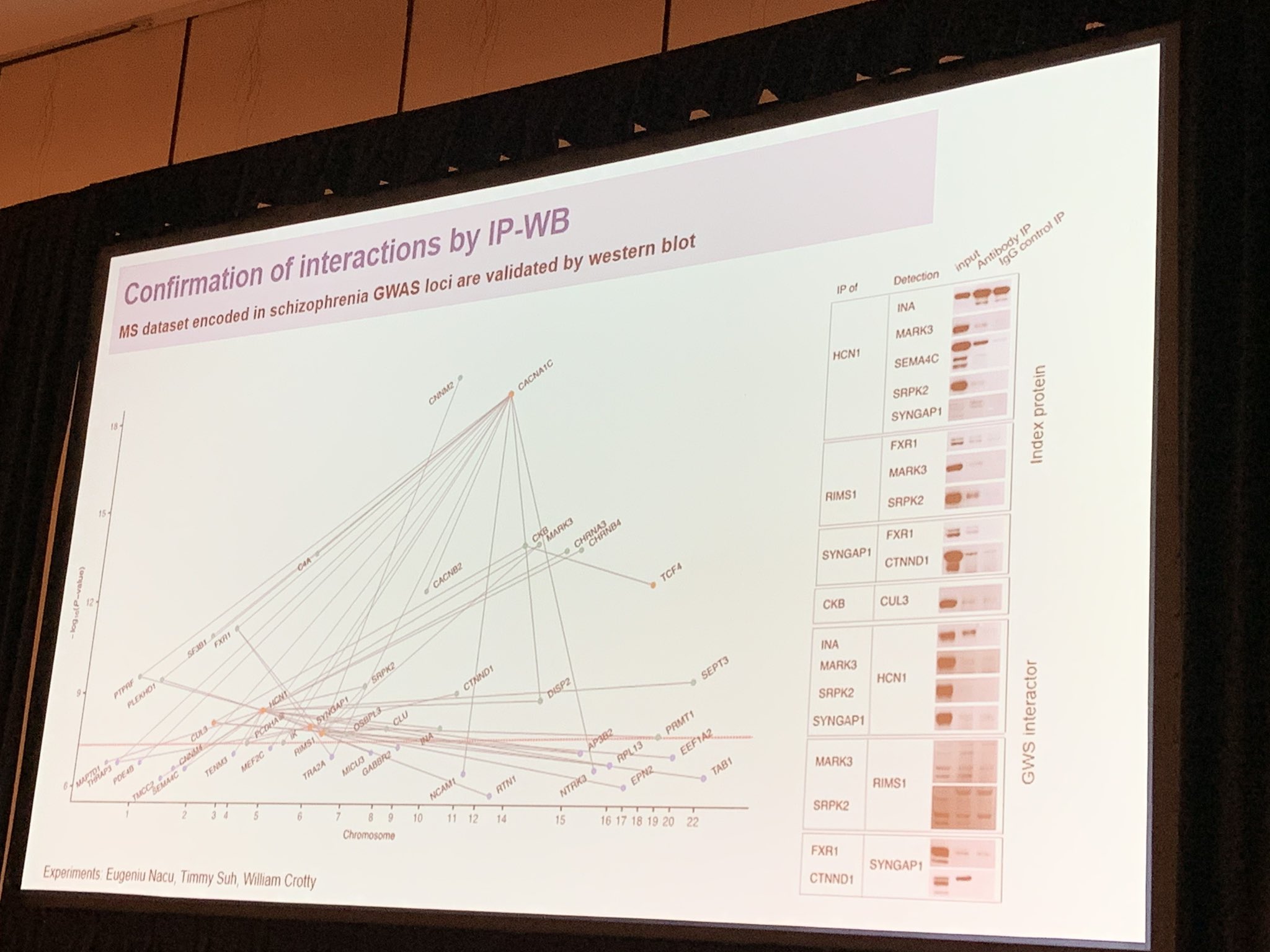
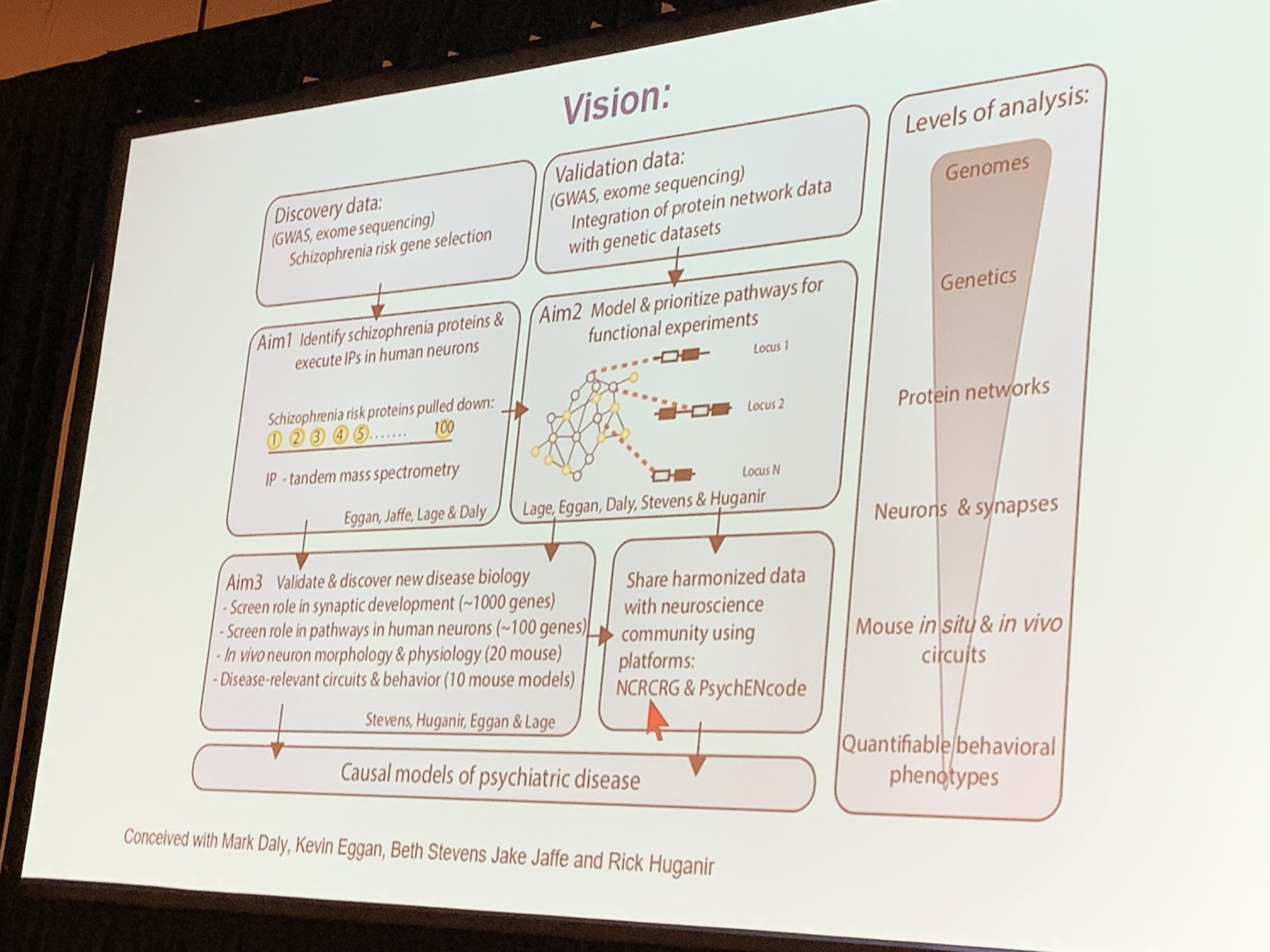
April presented the Stanley Center at the Broad Institute Brain Interaction Network (BINe) project, in collaboration with Kevin Eggan, Tarjinder Singh, Mark Daly and Hailiang Huang, to map neuron-specific protein interactions of schizophrenia genes.
Heiko presented his work on combining tumor genome data and protein interaction networks to identify tumor vulnerabilities, a project in great collaboration with James Neal and Gaddy Getz.




Kasper hosted a delegation led by Minister of Foreign Affairs of Denmark, Jeppe Kofod, at the Broad Institute to discuss world leading research and innovation going on in Denmark and Boston.
