From the lab Heiko Horn led the development and implementation of network-based statistic that identifies cancer driver genes with high accuracy from cancer genomes. Results are validated using a massively parallel in vivo tumorigenesis assay in mice and by re-analyzing 660 lung adenocarcinoma patients where ~1/3 do not have mutations or copy number changes in known oncogenes identifying two new cancer-driving genes underlying this cancer type.
This project is a collaboration with Jesse Boehm and Gad Getz from the Broad Institute and MGH Cancer Center.
Paper can be found here:
NetSig: network-based discovery from cancer genomes
Heiko Horn, Michael S Lawrence, Candace R Chouinard, Yashaswi Shrestha, Jessica Xin Hu, Elizabeth Worstell, Emily Shea, Nina Ilic, Eejung Kim, Atanas Kamburov, Alireza Kashani, William C Hahn, Joshua D Campbell, Jesse S Boehm, Gad Getz & Kasper Lage
Abstract:
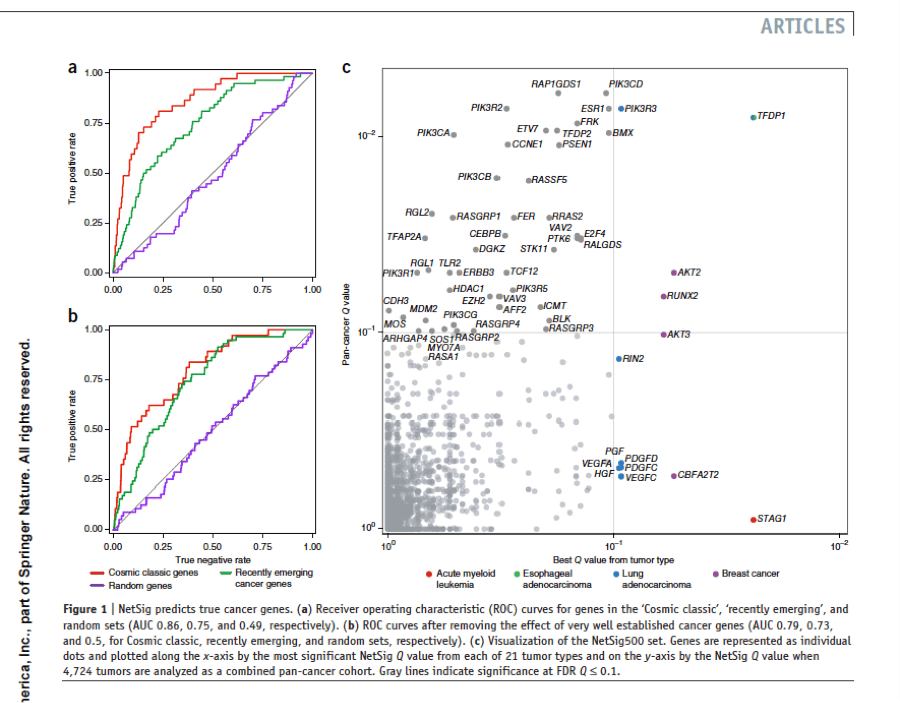
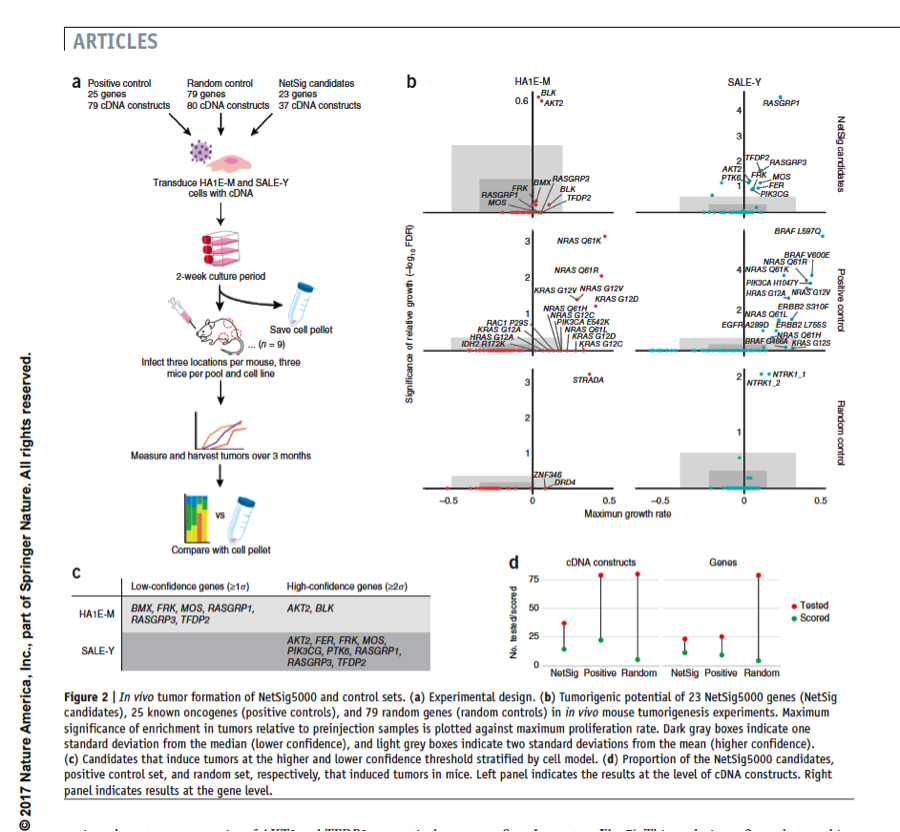
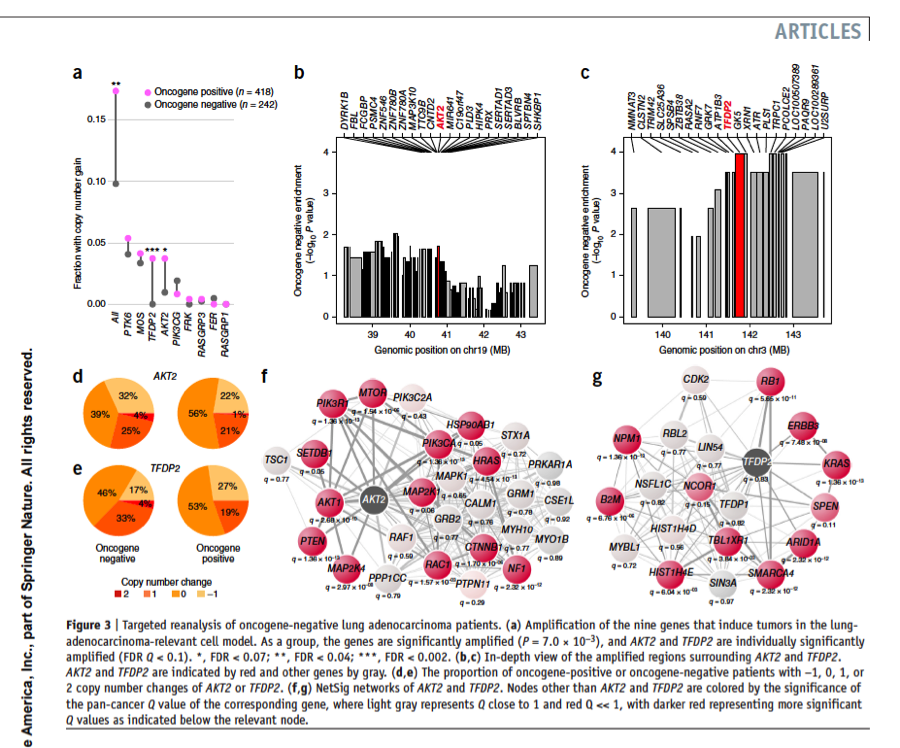
Methods that integrate molecular network information and tumor genome data could complement gene-based statistical tests to identify likely new cancer genes; but such approaches are challenging to validate at scale, and their predictive value remains unclear. We developed a robust statistic (NetSig) that integrates protein interaction networks with data from 4,742 tumor exomes. NetSig can accurately classify known driver genes in 60% of tested tumor types and predicts 62 new driver candidates. Using a quantitative experimental framework to determine in vivo tumorigenic potential in mice, we found that NetSig candidates induce tumors at rates that are comparable to those of known oncogenes and are ten-fold higher than those of random genes. By reanalyzing nine tumor-inducing NetSig candidates in 242 patients with oncogene-negative lung adenocarcinomas, we find that two (AKT2 and TFDP2) are significantly amplified. Our study presents a scalable integrated computational and experimental workflow to expand discovery from cancer genomes.